Синдром аперта
Содержание:
ÐбÑие ÑведениÑ

ÐеÑвÑй ÑлÑÑай данного наÑÑÑÐµÐ½Ð¸Ñ Ð±Ñл опиÑан еÑе в 1880 годÑ. СиндÑом ÐоÑаÑа — паÑологиÑ, пÑи коÑоÑой Ñ Ñеловека наÑÑÑаеÑÑÑ Ð²Ð¾ÑпÑиÑÑие вÑемени, он наÑÐ¸Ð½Ð°ÐµÑ Ð¾ÑÑиÑаÑÑ ÑебÑ. То еÑÑÑ Ð±Ð¾Ð»Ñной ÑÑиÑÐ°ÐµÑ ÑÐµÐ±Ñ ÑмеÑÑим. ÐÑи ÑÑом он Ð¼Ð¾Ð¶ÐµÑ Ð¾ÑвеÑгаÑÑ ÑолÑко некоÑоÑÑе ÑаÑÑи Ñвоего Ñела. ÐаÑÐ¸ÐµÐ½Ñ Ð¾ÑÑиÑÐ°ÐµÑ Ñвое ÑÑÑеÑÑвование, ÑÑиÑÐ°ÐµÑ ÐµÐ³Ð¾ непÑодÑкÑивнÑм и беÑÑмÑÑленнÑм. ÐекоÑоÑÑе лиÑа даже пеÑеÑÑаÑÑ Ð¿Ð¸ÑаÑÑÑÑ, Ñак как ÑÑиÑаÑÑ ÑÑо лиÑним, Ð²ÐµÐ´Ñ Ð¾Ð½Ð¸ же «Ð¼ÐµÑÑвѻ.
СиндÑом ÐоÑаÑа пÑÐµÐ´Ð¿Ð¾Ð»Ð°Ð³Ð°ÐµÑ Ð½ÐµÑколÑко ÑниженнÑй глÑкознÑй меÑаболизм и заÑоÑможеннÑÑ Ð´ÐµÑÑелÑноÑÑÑ Ð½ÐµÐºÐ¾ÑоÑÑÑ ÑÑаÑÑков мозга. То еÑÑÑ Ñеловек Ñловно наÑодиÑÑÑ Ð² вегеÑаÑивном ÑоÑÑоÑнии. СледÑÐµÑ Ð¾ÑмеÑиÑÑ, ÑÑо Ð´Ð°Ð½Ð½Ð°Ñ Ð±Ð¾Ð»ÐµÐ·Ð½Ñ ÑаÑе вÑего Ð²Ð¾Ð·Ð½Ð¸ÐºÐ°ÐµÑ Ñ Ð¶ÐµÐ½Ñин. ÐÑи ÑÑом ÑимпÑÐ¾Ð¼Ñ Ð¼Ð¾Ð³ÑÑ Ð¿Ð¾ÑвлÑÑÑÑÑ ÑпонÑанно. УÑÑиÑе, ÑÑо Ñ Ð¼Ð¾Ð»Ð¾Ð´ÑÑ Ð»Ñдей заболевание ÑазвиваеÑÑÑ Ñедко. Ðно более ÑаÑакÑеÑно Ð´Ð»Ñ Ð´ÐµÐ¿ÑеÑÑивнÑÑ ÑаÑÑÑÑойÑÑв в ÑÑаÑÑеÑком возÑаÑÑе.
ТеÑение Ñакого наÑÑÑÐµÐ½Ð¸Ñ Ð´Ð¾ÑÑаÑоÑно ÑÑжелое. ÐÑложнÑеÑÑÑ Ð¾Ð½Ð¾ Ñем, ÑÑо паÑÐ¸ÐµÐ½Ñ Ð½Ðµ обÑаÑаеÑÑÑ Ðº докÑоÑÑ, Ñак как ÑеÑÑÐµÑ ÑвÑÐ·Ñ Ñ ÑеалÑноÑÑÑÑ, и ÑвеÑен, ÑÑо ÐµÐ¼Ñ Ñже ниÑем нелÑÐ·Ñ Ð¿Ð¾Ð¼Ð¾ÑÑ.
Диагностика и симптомы болезни в Израиле
При синдроме Апера у детей наблюдается краниосиностоз — преждевременное сращение продольных и поперечных черепных швов. В результате этого по мере роста головного мозга череп деформируется, у детей развивается постоянно повышенное внутричерепное давление, что приводит к умственной отсталости, частым головным болям, тошноте и рвоте. На фоне деформации костей черепа у детей отмечаются типичные симптомы синдрома Аперта: «башнеобразная» форма головы с выпуклым и высоким лбом, выпячивание глазных яблок и увеличение расстояния между глазницами, вогнутая или плоская форма лицевого отдела, недоразвитие челюсти, приводящее к нарушению прикуса. Также синдром Аперта может сопровождаться потерей слуха, косоглазием, обструктивным апноэ сна, рецидивирующими инфекционными болезнями лор-органов.
Кроме того, при данном синдроме у ребенка после рождения нередко наблюдается синдактилия — срастание пальцев на нижних или верхних конечностях. В большинстве случаев пальцы (второй, третий и четвертый) срастаются в виде перепонок или полностью. Иногда у детей с синдромом Аперта обнаруживают врожденный порок сердца, а также аномалии развития позвонков, мочевыводящих путей, желудочно-кишечного тракта. Практически в каждом случае, у пациентов с акроцефалосиндактилией наблюдается карликовый рост.
Первичный диагноз можно установить после выявления синдактилии, характерных особенностей черепа и внешнего вида лица. Чтобы дифференцировать синдром Аперта от других врожденных генетических аномалий, израильские специалисты включают в диагностический курс следующие мероприятия:
- изучение семейного анамнеза, анализ жалоб;
- неврологический осмотр, оценка умственных способностей;
- молекулярно-генетический анализ крови, консультация генетика;
- офтальмологический осмотр глазного дна;
- компьютерная и магнитно-резонансная томография (КТ и МРТ) на предмет гидроцефалии, а также для исследования патологических особенностей головного мозга и черепа;
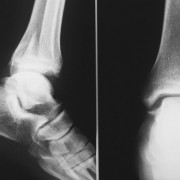
- рентгенография черепа и конечностей при синдактилии.
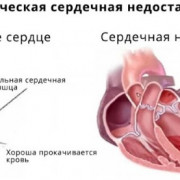
Поскольку при синдроме Аперта наблюдается широкий спектр нарушений, израильские врачи проводят всестороннее обследование. Поэтому пациентам могут назначить с основными диагностическими процедурами УЗИ почек и других органов, а также ЭхоКГ на наличие или отсутствие пороков сосудов и сердца, вызывающих сердечную недостаточность.
Для получения более точной информации о стоимости лечения и специальных предложениях нажмите кнопку
Преимущества МРТ диагностики
Рентгенография, принцип работы которой основан на способности тканей организма с разной степенью противостоять пропусканию рентгеновских лучей. В результате возможно проецировать изображение определённой части тела человека на черно – белый снимок. Этот способ хорошо позволяет диагностировать состояние скелета, некоторых органов. Но во время процедуры пациент получает, хоть и малую, но порцию радиации и исследование не получается полным.
В отличии от рентгенографии, магниторезонансная томография (МРТ) – это точный современный метод сканирования любого участка организма человека при помощи усиленных магнитных полей и радиочастотных импульсов. После обработки на компьютере получают изображение высокого качества, по которому можно судить о состоянии исследуемого не облучая его радиацией. Такой подход помогает обнаружить развитие болезни на ранней стадии, что зачастую может спасти жизнь человеку.
Установка состоит из специальной кушетки-платформы, расположенной внутри цилиндрической трубы — МРТ сканера. Пациент ложится таким образом, что ноги или голова находятся в центре аппарата. Процедура длится от 20 минут до часа (в особых случаях).
Профилактические рекомендации
Поскольку основной фактор развития синдрома – генные мутации, эффективной профилактики не существует. В данном контексте можно говорить о своевременной диагностике – УЗИ беременных женщин, особенно если в роду были случаи патологии.
Учитывая менее вероятные, но также существующие причины, к мерам профилактики можно отнести:
- недопущение рентгеновского облучения при беременности;
- своевременное лечение болезней при вынашивании плода.
Синдром Апера – редкая тяжёлая врождённая аномалия, не поддающаяся патогенетической терапии. Своевременное хирургическое и медикаментозное лечение носит паллиативный характер и может существенно улучшить качество жизни пациента.
Загрузка…
Патофизиология
Ихтиоз Арлекина сопровождается нарушениями микроциркуляции и периферического кровотока вследствие неврологических нарушений, что заключаются в одностороннем повреждении либо асимметрии функций нервной симпатической системы. И, как упоминалось выше, является патогномоничным признаком асфиксии либо ЧМТ новорожденных.
Существует предположение, что характерное для данной патологии утолщение (индурация) рогового слоя развивается вследствие дефекта синтеза липидов либо дефосфорилирования белков, в результате чего происходит их секреция в кожу и, как следствие, формирование последней нарушается.
Прогноз и профилактика
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения. При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов. В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Загрузка…
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности – к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей – различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Лечение[править | править код]
Радикальных методов лечения не существует.
Симптоматическое лечение данной патологии заключается в хирургическом увеличении объёма черепа, коррекция синдактилии и полидактилии.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Апера часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.
Консервативные методы лечения применяют для разработки суставов. Также, для стимуляции психического развития назначают ноотропные препараты: аминалон, пирацетам и другие, использовались в эпоху развития науки до проверки лечения методами доказательной медицины.
Симптоматические проявления
Изменения в костях предплечья выражаются:
- Атрофией области кисти и предплечья, в редком сочетании регистрируется совмещение с гипертрофическими изменениями локтевого сочленения;
- Нарушением двигательной способности плеча – при патологии наблюдаются проблемы с самостоятельным обслуживанием и снижение работоспособности;
- Ограниченностью свободных движений кистью.
Черепная аномалия представлена:
- Изменениями формы черепной коробки;
- Задержкой в психическом и интеллектуальном развитии малыша;
- Постоянно присутствующими мигренями;
- Тошнотой с переходом в рвоту;
- Судорожными состояниями;
- Непродолжительными потерями сознания;
- Нарушениями цикличности сна и бодрствования;
- Постоянной раздражительностью;
- Необоснованным беспокойством;
- Косоглазием;
- Выпиранием глазного яблока за пределы глазницы — экзофтальмом.
Патология Клиппеля-Фейля может сочетаться с иными поражениями и проявляться:
- Укороченным шейным отделом;
- Нарушением подвижности верхних участков позвоночного столба;
- Недостаточным ростом волосяного покрова или их отсутствием в затылочной области;
- Расщеплением позвоночника;
- Понижением уровня чувствительности кожных покровов;
- Приподнятым положением линии лопаток и присутствием крыловидной складчатости;
- Сколиозом.
Данный тип патологического отклонения может сопровождаться:
- Разнообразными пороками сердца;
- Отклонениями в развитии мочеполового отдела;
- Волчьей пастью – расщеплением тканей твердого и мягкого неба;
- Аномалиями пальцев нижних и верхних конечностей;
- Изменениями в ребрах и почках;
- Отклонениями в органах дыхания;
- Изменениями лица.
При любом подвиде заболевания присутствуют постоянные, ноющие боли. Основным источником болезненных ощущений является место деформации костных тканей.
Что такое синдром Аперта?
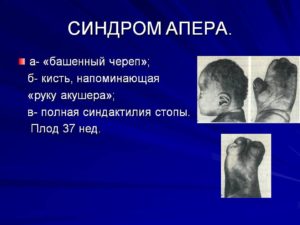
Синдром Аперта — редкое генетическое заболевание, которое проявляется аномалиями лица и черепа, а также нарушениями зрения и стоматологическими проблемами. Синдром Аперта также может вызвать аномалии пальцев рук и ног.
Википедия
Симптомы синдрома Аперта
Характерные симптомы синдрома Аперта у детей:
- конусообразный череп — туррибрахицефалия;
- лицо с углублением посередине;
- глаза широкие и выпуклые наружу;
- клювовидный нос;
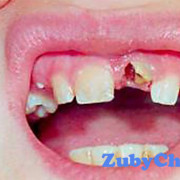
- недоразвитая верхняя челюсть, которая может привести к скученности зубов.
Аномалии лица и черепа могут привести к некоторым проблемам со здоровьем и развитием. Проблемы со зрением зачастую являются результатом маленьких глазниц.
Верхняя челюсть обычно меньше, чем нижняя, что может привести к проблемам с зубами. При синдроме Аперта череп меньше обычного, что может оказать давление на развивающийся головной мозг.
У людей с синдромом Аперта может быть средний уровень интеллекта или слабые или умеренные интеллектуальные нарушения.
https://www.youtube.com/watch?v=W04Jmp00V0w
Дети с синдромом Аперта зачастую имеют перепончатые пальцы рук и ног. Чаще всего три пальца ног срастаются вместе (синдактилия), но иногда все пальцы могут быть перепончатыми. Реже у ребенка могут быть дополнительные пальцы.
Дополнительные признаки и симптомы синдрома Аперта
- потеря слуха;
- угри;
- сильное потоотделение;
- слияние шейных позвонков;
- жирная кожа;
- отсутствие бровей;
- задержки роста и развития;
- рецидивирующие инфекции уха;
- волчья пасть;
- легкое или умеренное снижение интеллекта.
Причины синдрома Аперта
Синдром Аперта — генетическое состояние, вызванное мутацией в гене FGFR2. Это может произойти как у детей, не имеющих в семейном анамнезе данного синдрома, так и передаться по наследству от родителей.
Ген FGFR2 продуцирует белок, называемый рецептором фактора роста фибробластов. Этот белок играет важную роль в развитии плода, включая ключевую роль в сигнальном развитии костной клетки.
Когда происходит мутация этого гена, FGFR2 сигнализирует дольше, чем обычно, что приводит к раннему слиянию костей черепа, лица, пальцев стоп и рук.
Мутации в гене FGFR2 также могут вызвать ряд других сопутствующих нарушений:
- Синдром Пфайффера;
- Синдром Крузона
- Синдром Джексона-Вейсса.
Распространенность синдрома Аперта
Синдром Аперта — крайне редкое заболевание. Количество людей, которые имеют его, не известно.
Большинство случаев синдрома Аперта появляются без наличия в семейном анамнезе данного синдрома.
Тем не менее, когда один из родителей имеет данное расстройство, у ребенка будет 50-процентная вероятность его развития. Этот синдром встречается как у мужчин, так и у женщин в равной степени.
Диагностика синдрома Аперта
Википедия
Синдром Аперта зачастую можно диагностировать при рождении или в раннем возрасте. Чтобы официально диагностировать синдром Аперта, врач должен найти характерные аномалии костей головы, рук и ног. Врач может назначить рентгенографию черепа или КТ головы, чтобы определить характер аномалий кости. Для диагностики может быть использовано молекулярно-генетическое тестирование.
Лечение синдрома Аперта
План лечения будет составлен с учетом индивидуальных особенностей и может включать в себя хирургическое лечение, чтобы уменьшить давление на головной мозг, изменить черты лица или осуществить разделение пальцев.
Ребенку с синдромом Аперта потребуются пожизненные наблюдения. Врач проверит наличие дополнительных осложнений, вызванных синдромом Аперта, и предложит соответствующие методы лечения.
Общие методы лечения осложнений, связанных с синдромом Аперта
- исправление проблем со зрением;
- лечение задержки развития и роста;
- стоматологические процедуры для коррекции скученности зубов.
Ранняя диагностика у младенцев улучшит шансы успешного лечения некоторых симптомов и осложнений синдрома Аперта.
Синдром Аперта не влияет на продолжительность жизни человека, хотя проблемы с сердцем или другие связанные с ним состояния могут привести к осложнениям. Лечение может улучшить внешний вид человека с синдромом Аперта.
Литература
Robin N. H., Falk M. J., Haldeman-Englert C. R. FGFR-related craniosynostosis syndromes. GeneReviews. – 2011.
Читайте нас в Telegram – Medical Insider
Лечение
Лечение синдрома Аперта варьируется в зависимости от того, какие симптомы наблюдаются у больного. Лечение может потребовать ухода со стороны группы медицинских специалистов, включая педиатров и хирургов, нейрохирургов, врачей, специализирующихся на заболеваниях скелета, суставов и мышц (ортопедов), врачей, специализирующихся на заболеваниях ушей, носа и горла (отоларингологов), врачей, специализирующихся на нарушениях сердечной деятельности (кардиологов).
Специальные методы лечения синдрома Аперта являются симптоматическими и поддерживающими. Краниосиностоз и гидроцефалия могут привести к ненормально повышенному давлению внутри черепа и мозга. В таких случаях для коррекции краниосиностоза может быть рекомендовано раннее хирургическое вмешательство (в течение 2-4 месяцев после рождения). Для пациентов с гидроцефалией операция может также включать введение трубки (шунта) для оттока избыточной спинномозговой жидкости (СМЖ) из головного мозга. СМЖ сливается в другую часть организма, где он абсорбируется.
Корректирующая и реконструктивная хирургия может быть рекомендована для коррекции черепно-лицевых пороков развития. Хирургия также может помочь исправить полидактилию и синдактилию, а также другие скелетные дефекты или физические отклонения. Для больных с врожденными пороками сердца может потребоваться лечение определенными препаратами, хирургическое вмешательство и/или другие терапевтические меры. Для некоторых больных с нарушениями слуха полезны слуховые аппараты.
Раннее вмешательство может быть важным для обеспечения того, чтобы дети с синдромом Аперта полностью раскрыли свой потенциал. Специальные терапевтические мероприятия, такие как физиотерапия, трудотерапия и специализированное обучение также будут полезными.
Для пострадавших лиц и их семей рекомендуется генетическое консультирование. Генетический консультант сможет объяснить причины заболевания. Он также может обсудить возможность рождения новых детей с заболеванием. Дополнительно, для всей семьи необходима психосоциальная поддержка.
Лечение
Лечение расстройства варьируется в зависимости от конкретных симптомов. Операция на мозге помогает уменьшить давление, создаваемое краниосиностозом. Команда врачей необходима для работы с ребенком с синдромом Аперта, чтобы планировать операции, лечить лицевые аномалии, слияние пальцев рук или ног.
Стандартное лечение включает раннюю краниэктомию (хирургическое удаление части черепа) и косметическую реконструкцию, чтобы способствовать нормальному росту лица.

Многодисциплинарная помощь предусматривает оценку и лечение симптомов:
- Офтальмологическое лечение амблиопии, аметропии, косоглазия;
- Аудиологическую помощь, миринготомию при потере слуха;
- Шунтирование для снятия внутричерепного давления из-за гидроцефалии (увеличение цереброспинальной жидкости в черепной полости);
- Трахеостомия (разрез гортани) для устранения обструкции дыхательных путей;
- Ортодонтическая хирургия для коррекции нарушений зубов, челюсти.
Люди, страдающие от расстройства, должны тщательно наблюдаться на протяжении всей жизни.
Причины возникновения синдрома Апера
Симптомокомплекс, характерный для синдрома, впервые описал французский педиатр Эжен Апер (Аперт) в начале XX века. Аномалия также носит названия акрокраниодисфалангия, акроцефалосиндактилия, акросфеносиндактилия – все эти слова в переводе с латыни характеризуют отклонения от нормы в строении черепа, фаланг пальцев, описывая внешние признаки патологии.
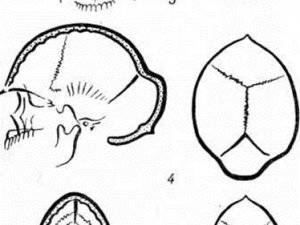
Череп плода, находящегося в утробе матери, состоит из частей, которые ещё не полностью срослись между собой. Между теменными и лобными костями находится коронарный (венечный) шов, а между теменными – стреловидный (сагиттальный). Заращение этих соединительнотканных участков при описываемой аномалии происходит ранее, чем положено. Это приводит к появлению внешних признаков болезни и развитию внутренних патологий.

Мальчики с синдромом Апера рождаются также часто, как и девочки, то есть заболевание одинаково распространено среди представителей обоих полов. Основными факторами развития патологии считаются:
- Генная мутация. Изменение наследственных свойств в клетках организма одного из родителей в данном случае носит спонтанный характер. Ген, находящийся на десятой хромосоме и называемый FGFR2, расположен таким образом, что легко подвергается повреждениям. Именно этот участок ДНК отвечает за формирование и развитие соединительной (в частности, костной) ткани в эмбриональном периоде. Дефект гена приводит к заболеваниям костной системы. Вероятность рождения ребёнка с описываемым симптомокомплексом у пожилых родителей выше, чем у молодых.
- Наследственность. У человека, страдающего синдромом, с вероятностью 50% родится ребёнок с такой же аномалией.
- Рентгеновское облучение беременной женщины.
- Заболевания, перенесённые беременной: вирусы гриппа, туберкулёз, сифилис, краснуха и некоторые другие.
Синдром Апера, причины которого довольно сложно установить при отсутствии генетической предрасположенности, практически не поддаётся лечению.
Признаки и симптомы
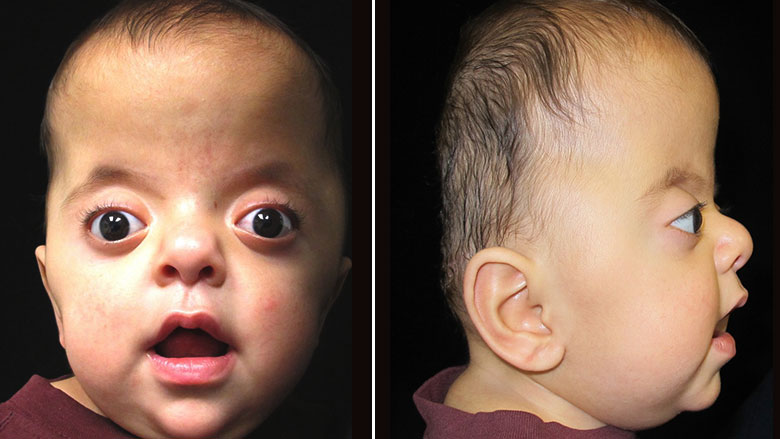
Синдром Аперта характеризуется краниосиностозом, преждевременным закрытием фиброзных суставов (швов) между определенными костями черепа. У детей без краниосиностоза швы позволяют голове расти и расширяться. Затем, эти кости сливаются воедино, образуя череп.
У людей с краниосиностозом мозг все еще растет после преждевременного закрытия этих швов.
Возникающее давление при росте головного мозга может привести к тому, что различные кости черепа и лица деформируются во время развития.
В зависимости от того, какие швы преждевременно закрываются, варьируется степень их тяжести. У большинства больных людей происходит преждевременное закрытие швов между костями, образующими лоб и верхнюю часть черепа. Это приводит к тому, что голова с рождения становится заостренной на верхней части (акроцефалия). Вдобавок, задняя часть черепа может выглядеть сплющенной, с высоким и широким лбом (см. фото выше). На черепе может быть большой «родничок» с поздним закрытием.
У некоторых больных также может быть гидроцефалия, при которой в полостях головного мозга накапливается спинномозговая жидкость. Это может вызвать внутричерепное давление.
Лицевые кости тоже могут быть поражены краниосиностозом. Это может привести к характерным дефектам лица. У пациентов с синдромом Аперта часто наблюдаются широко расставленные глаза (гипертелоризм), выпученные глаза (экзофтальм) или наклоненные вниз глазные щели. У них также могут быть слаборазвитые срединно-лицевые области (гипоплазия верхней челюсти) и расщелина нёба. Правая и левая стороны лица могут быть не симметричными.
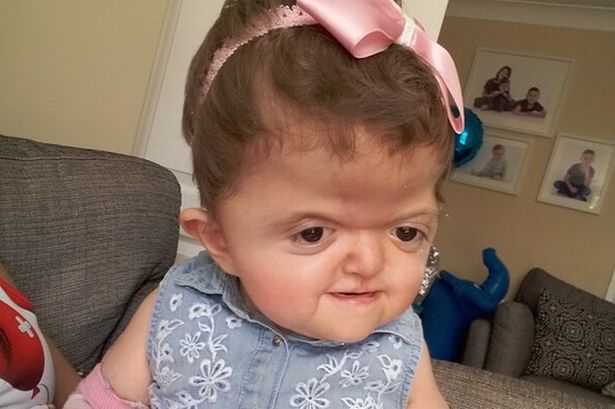
Люди с болезнью могут иметь сплющенный нос с низкой переносицей. У людей бывает задержка роста зубов, скученность зубов или открытый прикус.
Если отверстия между носом и горлом сужены или заблокированы, или трахеальный хрящ поврежден, это может помешать дыханию и глотанию. Люди с этими проблемами могут иметь инфекции верхних дыхательных путей, апноэ во сне, недоедание.
Синдром Аперта имеет несколько характерных пороков развития кисти и стопы. У пострадавших людей могут быть короткие, широкие и большие пальцы ног, отклоняющиеся наружу. Они также могут иметь частичное или полное слияние (синдактилию) определенных пальцев рук и ног. Многие больные люди имеют полное слияние костей второго и четвертого пальцев и один единственный сплошной ноготь («варежкообразная» синдактилия). Однако возможны и другие слияния.
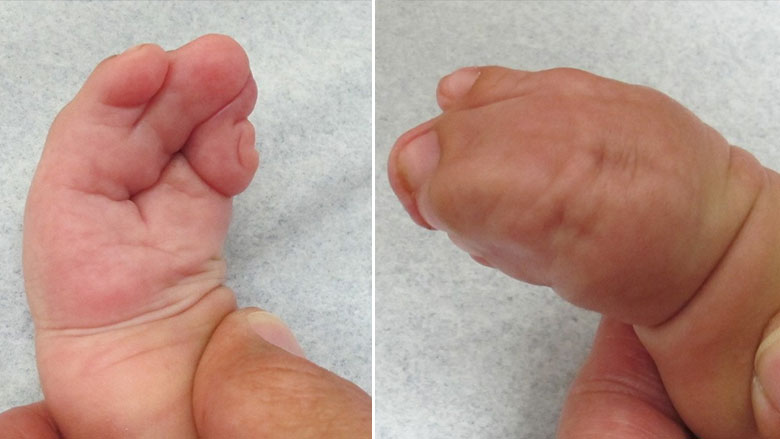
Суставы пальцев становятся жесткими к четырем годам. В ногах синдактилия также обычно затрагивает вторые, третьи и четвертые пальцы. Ногти на ногах могут быть частично непрерывными или отдельными. Как правило, синдром поражает верхние конечности сильнее, чем нижние.
Синдром Аперта может влиять и на другие системы органов (см. таблицу).
| Скелетная система |
|
| Нервная система |
|
| Уши |
|
| Сердечно-сосудистая система | |
| Брюшная полость |
|
| Почки и мочеполовая система |
|
Диагностирование заболевания
Для постановки диагноза требуются следующие мероприятия:
Врач должен проанализировать жалобы пациента, а также анамнез болезни
Необходимо выяснить, имелись ли в семье случаи развития подобной патологии;
Неврологическое обследование, чтобы оценить форму черепа и интеллектуальное развитие пациента (специальные анкеты, а также беседа);
Обследование глазного дна, чтобы выявить наличие симптомов увеличения внутричерепного давления (отёк ДЗН, а также размытость его краёв);
Для оценки состояния черепа выполняется его рентгенография;
Компьютерная, а также магнитно-резонансная томография головы, чтобы послойно обследовать структуру головного мозга с черепом, определить наличие симптомов преждевременного сращения черепных швов, а помимо этого гидроцефалию (из-за увеличения внутричерепного давления скапливается излишек ликвора (это цереброспинальная жидкость, которая способствует процессу обмена веществ, а также питанию головного мозга));
Рентген стоп с кистями, чтобы узнать причину, из-за которой произошло сращение пальцев (это важно для планирования последующего хирургического вмешательства);
Может быть назначена консультация у медицинского генетика, а также нейрохирурга.